SQL Query Syntax
OmixAtlas class enables users to interact with functional properties of the omixatlas such as create and update an Omixatlas, get summary of it's contents, add, insert, update the schema, add, update or delete datasets, query metadata, download data, save data to workspace etc.
Parameters:
-
token(str, default:None) –token copy from polly.
Usage
from polly.OmixAtlas import OmixAtlas
omixatlas = OmixAtlas(token)
query_metadata
This function will return a dataframe containing the SQL response. In cases of data intensive queries, the data returned can be very large to process and it might lead to kernel failure , where in the runtime memory is exhausted and the process haults. In order to access the data, there are two options in this case, either increase the kernel memory, or use the function query_metadata_iterator() that returns an iterator. The function usage can be looked up in the documentation mentioned here under the Polly Python section - "https://docs.elucidata.io/index.html".
Parameters:
-
query(str) –sql query on omixatlas.
-
experimental_features–this section includes in querying metadata
.
Returns:
-
DataFrame–It will return a dataframe that contains the query response.
Raises:
-
UnfinishedQueryException–when query has not finised the execution.
-
QueryFailedException–when query failed to execute.
query_metadata_iterator
This function will return a Generator object containing the SQL response.
Parameters:
-
query (str)–sql query on omixatlas.
-
experimental_features–this section includes in querying metadata
.
Returns:
-
None–It will return a generator object having the SQL response.
Raises:
-
UnfinishedQueryException–when query has not finised the execution.
-
QueryFailedException–when query failed to execute.
Examples
query_metadata()
Querying the dataset level metadata:
query = "SELECT [column_name] FROM [repo_name/repo_id].datasets WHERE [column_name]='[value]'"
-
To identify datasets belonging to the tissue Breast, disease Breast Neoplasms and organism Homo sapiens
-
Fetch datasets from Depmap which has gene dependency information according to CRISPR screening experiments
-
Identify all transcriptome datasets in Hepatocellular Carcinoma disease in Human and Mouse
Querying the sample level metadata:
- For all samples except Single Cell
query = "SELECT [column_name] FROM [repo_name/repo_id].samples WHERE [column_name]='[value]'"
-
Get the name of samples, dataset ID and extract_protocol_ch1 where spectroscopy is mentioned in the extract_protocol_ch1
-
Get the name of disease and number of samples where information about particular disease is curated
-
For samples (cells) in Single Cell
query = "SELECT [column_name] FROM [repo_name/repo_id].samples_singlecell WHERE [column_name]='[value]'"
- Find all the sample level metadata for the dataset ID 'GSE141578_GPL24676'
query = "SELECT * FROM sc_data_lake.samples_singlecell WHERE src_dataset_id = 'GSE141578_GPL24676' "
cells = omixatlas.query_metadata(query)
Querying the feature level metadata:
- For all features except Single Cell
query = "SELECT [column_name] FROM [repo_name/repo_id].features WHERE [column_name]='[value]'"
- For features (genes) in Single Cell
query = "SELECT [column_name] FROM [repo_name/repo_id].features_singlecell WHERE [column_name]='[value]'"Querying data matrix
At the dataset level metadata, there's a column names data_table_name. This information will be helpful for users to run data matrix level queries. The template query looks as follow:-
Example: Fetch data matrix for selected genes for a dataset ID of interest
```
gene = ('hbb-y', 'fth1', 'bbip1', 'actb')
query = f"SELECT * FROM data_matrices.geo_transcriptomics_omixatlas__GSE143319_GPL20301_raw WHERE
LOWER(rid) IN {gene}"
```
Query specific to source and datatype in an OmixAtlas
If the schema of an OmixAtlas has multiple source and data types then querying for the specific source and/or datatype can be performed as shown below.
from polly import Omixatlas
omixatlas = Omixatlas()
query = """SELECT * FROM repo_name.{source_name_in_schema}.{datatype_name_in_schema}.datasets WHERE CONTAINS(curated_disease, 'Multiple Myeloma')
results-omixatlas.query_metadata(query)
results
get_schema function of an OmixAtlas to see the source_name and datatype_name to be used.
For clearer understanding of this feature, let’s look at a case:-
In the schema shown below for repo_id: 1659450268526, we can see there are 3 different sources. Out of these sources, the source lincs has 2 datatypes - mutation and transcriptomics
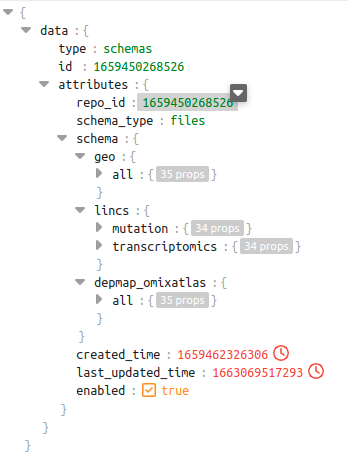
dataset level schema of repo_id 1659450268526
Query examples which will be supported in the above context:-
query = """SELECT * FROM 1659450268526.geo.datasets WHERE CONTAINS(curated_disease, 'Multiple Myeloma') """
query = """SELECT * FROM 1659450268526.lincs.datasets WHERE CONTAINS(curated_disease, 'Multiple Myeloma') """
query = """SELECT * FROM 1659450268526.lincs.transcriptomics.datasets WHERE CONTAINS(curated_disease, 'Multiple Myeloma') """The response to these queries will have a dataframe with columns which are specific to the source and datatype as per the schema shown above.
Other Query Examples
Select a few feature level metadata for selected genes from Mutation datasets of TCGA where dataset_id contains BRCA
```
query = """SELECT src_dataset_id, disease, protein_position, amino_acids, sequencer, impact, variant_class, consequence, name
FROM tcga.features AS features
JOIN (
SELECT dataset_id AS dataset_id, curated_disease AS disease FROM tcga.datasets WHERE data_type LIKE 'Mutation') AS datasets
ON features.src_dataset_id = datasets.dataset_id
WHERE hugo_symbol IN ('TP53','PIK3CA','CDH1','GATA3') AND features.src_dataset_id LIKE '%BRCA%'
ORDER BY features.src_dataset_id"""
```
Tutorial Notebooks
- Example queries have been given in various notebooks in this github folder.
- Sample queries for "geo" have been mentioned in the given notebook.
- Refer to this notebook for finding datasets of interest.
- For single cell queries, please check this notebook
query_metadata_iterator()
# Install polly python
pip install polly-python
# Import libraries
from polly.auth import Polly
from polly.omixatlas import OmixAtlas
# Create omixatlas object and authenticate
AUTH_TOKEN=(os.environ['POLLY_REFRESH_TOKEN'])
Polly.auth(AUTH_TOKEN)
omixatlas = OmixAtlas()
query = "SELECT * FROM sc_data_lake.samples_singlecell where src_dataset_id in ('Skin', 'SCP1402_nonda', 'GSE160756_GPL20795', 'GSE156793_GPL24676_Adrenal', 'Single-cell_RNA-seq_of_the_Adult_Human_Kidney_Version_1.0')"
iterator = omixatlas.query_metadata_iterator(query)
Implementation example
In the above execution of the query metadata function, a generator object is returned and stored in the variable "iterator". The python script given below uses the iterator to get the data one row at a time and appending it to a buffer list. The buffer limit set is 100 MB, after which the data is merged to the master dataframe. Repeating the same, we get the entire data in one dataframe.
# Sample Script for users
import pandas as pd
total_size=0
total_rows=0
total_df = pd.DataFrame()
result=[]
cumulative_size=0 # To calculate the size of the data collected iteratively
buffer_size=100 # Variable value in MB for buffer array, can be changed according to the need
for i in iterator:
result.append(i)
size = sys.getsizeof(i)
cumulative_size+=size
size_in_MB = cumulative_size/(1024*1024) # Converting bytes to MB
if size_in_MB >= buffer_size:
total_df = total_df.append(pd.DataFrame.from_records(result))
result.clear() # clearing records from the buffer for optimisation
total_size+=cumulative_size
cumulative_size=0
total_rows+=1
if size_in_MB < buffer_size:
total_size=cumulative_size
total_df = total_df.append(pd.DataFrame.from_records(result))
print(total_df.shape)
SQL Query Syntax
Complete Syntax
The complete syntax for searching and aggregating data is as follows:
[ WITH with_query [, ...] ]
SELECT [ ALL | DISTINCT ] select_expression [, ...]
[ FROM from_item [, ...] ]
[ WHERE condition ]
[ GROUP BY [ ALL | DISTINCT ] grouping_element [, ...] ]
[ HAVING condition ]
[ { UNION | INTERSECT | EXCEPT } [ ALL | DISTINCT ] select ]
[ ORDER BY expression [ ASC | DESC ] [ NULLS FIRST | NULLS LAST] [, ...] ]
[ OFFSET count [ ROW | ROWS ] ]
[ LIMIT [ count | ALL ] ]
Writing conditions with operators
The following operators can be used to define the conditions in the above mentioned queries:
| Operators | Functions performed |
|---|---|
= |
Equal to operator which can be used to find matching strings with values in the columns |
<> |
Not equal to operator which can be used to find non-matching strings with values in the columns |
> |
Greater than operator which can be used ONLY for integer based columns |
< |
Less than operator which can be used ONLY for integer based columns |
>= |
Greater than or equal to operator which can be used ONLY for integer based columns |
<= |
Less than or equal to operator which can be used ONLY for integer based columns |
IS NULL |
Check if the field value is NULL. |
IS NOT NULL |
Check if the field value is NOT NULL. |
AND |
All values across the parameters searched for have to be present in a dataset for it to be returned as a match when the AND operator is used. e.g. “organism = ‘Homo sapiens' AND disease = 'Carcinoma, Hepatocellular’” would only return datasets that belong to homo sapiens and have the disease as hepatocellular carcinoma. |
OR |
Atleast any one value across the parameters searched for have to be present in a dataset for it to be returned as a match when the OR operator is used. e.g. organism = 'Homo sapiens' OR disease = 'Carcinoma, Hepatocellular' would return datasets that belong to homo sapiens or have the disease as hepatocellular carcinoma or match both criteria. |
MATCH QUERY( |
It works like a fuzzy search. If you add a string for a parameter with this operator, it would return all possible results matching each word in the string. The search output is returned with a “Score” using which the output is sorted. e.g. MATCH_QUERY(description,'Transcriptomics profiling') would return all datasets having transcriptomics profiling , Transcriptomics and profiling as possible terms within their description. Each dataset would be scored on the basis of matching of the searched string with the information present within the dataset. |
MATCH PHRASE( |
This can be used for exact phrase match with the information being searched for. e.g. MATCH_PHRASE(description,'Transcriptomics profiling') would only return the datasets that have Transcriptomics profiling within their description. |
MULTI MATCH('query'='value', 'column_name'='value) |
This can be used to search for text in multiple fields, use MULTI MATCH('query'='value', 'column_name'='value). e.g. MULTI MATCH('query'='Stem Cells', 'fields'='tissue','description') would return datasets that have "Stem Cell" in either tissue OR description fields. |
GROUP BY |
The GROUP BY operator groups rows that have the same values into summary rows. The GROUP BY statement is often used with aggregate functions (COUNT, MAX, MIN, SUM, AVG) to group the result-set by one or more columns. |
HAVING |
Use the HAVING clause to aggregate inside each bucket based on aggregation functions (COUNT, AVG, SUM, MIN, and MAX). The HAVING clause filters results from the GROUP BY clause |
COUNT(*) |
This counts each row present in a table/index being queried. NOTE: The output of this query would return a JSON stating the total number of rows in the table |
LIMIT |
NOTE: The response of any query returns 200 entries by default. You can extend this by defining the LIMIT of the results you want to query to be able to return. |
ORDER BY |
Can only be used to sort the search results using integer based parameters in the schema. Sorting on the basis of dataset_id, number of samples, _score of the data is available at the dataset-level metadata. ASC or DESC can be used to define whether you want to order the rows in ascending or descending order respectively |